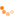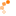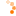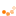Das im Jahr 2006 entwickelte Diketiminatosilylen zeigt aufgrund seiner zwitterionischen
Struktur im Gegensatz zu gewöhnlichen Silylenen eine bemerkenswerte Reaktivität. Neben den
beiden reaktiven Funktionen am Siliciumzentrum verfügt das Diketiminatosilylen über eine
zusätzliche Donorfunktion im Ligandenrückgrat. Ziel dieser Doktorarbeit ist es die Reaktivität
und katalytische Aktivität des ?-Diketiminatosilylens und dabei insbesondere dessen
Eigenschaften als Ligand in Übergangsmetallkomplexen zu untersuchen. Die Umsetzung des Silylens
mit Arsan liefert beispielsweise durch eine doppelte As-H-Bindungsaktivierung ein
donorstabilisiertes Arsasilen mit einer einzigartigen HSi=AsH-Einheit. Die Reaktion des
Silylens mit Ammin als Wasserstoffquelle verläuft hingegen zum thermodynamisch stabilen 1
1-Additionsprodukt. Durch gezielte Koordination des Silylens an ein Nickelzentrum lässt sich
die Donorfunktion am Siliciumzentrum schützen. Die Wasserstoffaddition mit Aminboran erfolgt
nun nur selektiv am Silylenliganden zu dem entsprechenden Hydridosilylen-Nickel-Komplex
während das Nickeltricarbonyl-Molekülfragment unangetastet bleibt. Der Si(II)hydrid-Komplex
wird anschließend erfolgreich in Hydrosilylierungsreaktionen mit Alkinen ohne Zusatz eines
exogenen Katalysators getestet. Die stöchiometrische Hydrosilylierung des Si(II)hydrids mit
Diphenylacetylen liefert chemoselektiv das Hydrosilylierungsprodukt dessen Alkenyleinheit
cis-konfiguriert vorliegt. Es konnte gezeigt werden dass das Nickeltricarbonyl-Fragment im
Si(II)hydrid-Nickelkomplex nicht nur eine Schutzgruppenfunktion besitzt sondern in der
Hydrosilylierungsreaktion eine entscheidende Rolle spielt. Darüber hinaus wurde eine neue
Methode für die Synthese neuartiger Übergangsmetallkomplexe basierend auf dem
Diketiminatosilylen entwickelt. Da das freie Silylen mit den Chlor-verbrückten Dimerkomplexe
unter milden Bedingungen keine Reaktion eingeht wurde zunächst die Reaktion des Silylens mit
HCl bei tiefen Temperaturen durchgeführt. Hierbei findet eine 1 4-Addition von HCl an das
Silylen statt wobei als labiles Intermediat ein Chlorsilylen entsteht. Dieses ist in der Lage
den Metallkomplex (Metall = Rh Ir) aufzubrechen und an das entsprechende Metallzentrum zu
koordinieren. Die anschließenden Untersuchungen der katalytischen Aktivität beider Komplexe
zeigten dass die katalytische Reduktion von Amiden mit dem Chlorsilylen-Rhodiumkomplex als
Präkatalysator chemoselektiv verläuft und der Zusatz an Li-Superhydrid die katalytische
Aktivität des Komplexes hemmt. Der Chlorsilylen-Iridiumkomplex zeigt im Vergleich zum analogen
Rhodiumkomplex eine deutlich höhere katalytische Aktivität und eine veränderte
Chemoselektivität.
")